Какой металл накапливается в органах человека при болезни вильсона
Обновлено: 09.05.2024
Болезнь Вильсона — Коновалова (гепатолентикулярная дегенерация, болезнь Вестфаля — Вильсона) — редкое наследственное заболевание, нарушение метаболизма меди, характеризующееся ее избыточным накоплением в тканях и жизненно-важных органах (в основном в печени, почках, головном мозге, глазах).
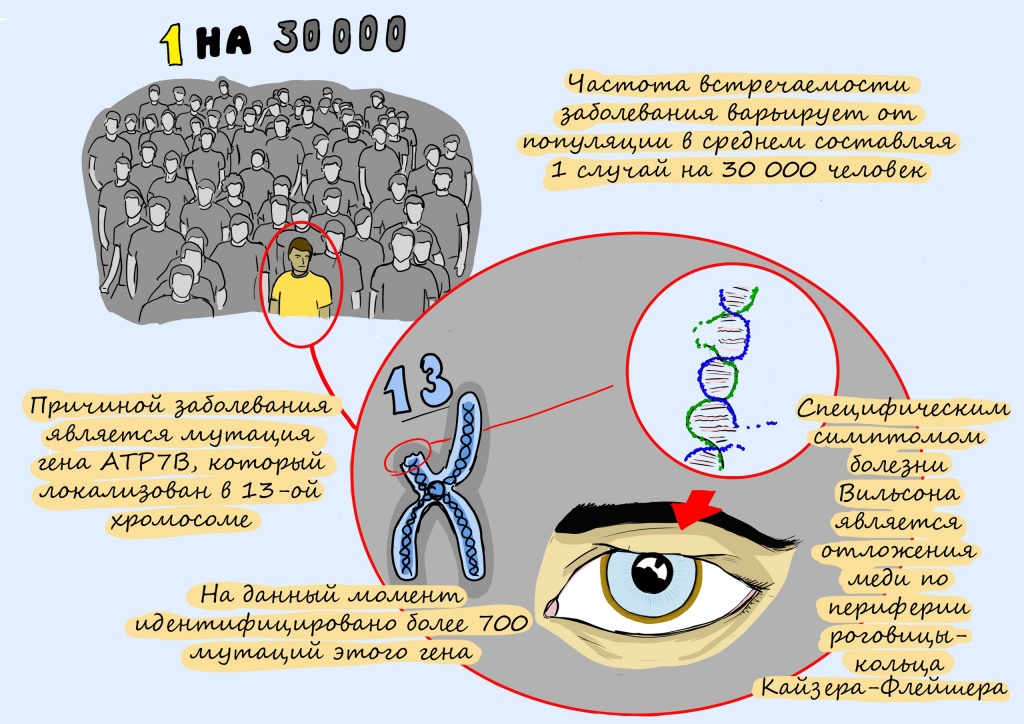
Распространенность болезни Вильсона — Коновалова во всем мире составляет 1 случай на 30 000–100 000 человек. Данные приблизительные, поскольку пациенты с гепатолентикулярной дегенерацией иногда получают ошибочный диагноз (в этот список входят различные болезни печени, неврологические заболевания и психические расстройства). Оценка носительства гена заболевания колеблется, однако считается, что его носителем может быть 1 человек из 90.
Причины возникновения
Болезнь Вильсона — Коновалова наследуется по аутосомно-рецессивному типу (мутации гена — в данном случае ATP7B — наследуются от обоих родителей). При таком типе наследования у каждого ребенка носителей дефектного гена существует 25% вероятность заболеть и 50% вероятность носительства мутации.
Ген ATP7B отвечает за метаболизм меди и играет важную роль в выведении избытка меди из организма. На сегодняшний день выявлено более 300 мутаций гена ATP7B.
Симптомы
Возраст манифестации заболевания варьируется: симптомы могут начать проявляться у ребенка 3–5 лет или же болезнь не дает о себе знать десятилетиями. Однако чаще всего болезнь Вильсона — Коновалова развивается к сорока годам, в редких случаях — после пятидесяти лет. Клиническая картина заболевания зависит от пола и возраста, так, у детей (средний возраст — 10 лет) чаще преобладают поражения печени.
К признакам и симптомам заболевания относятся:
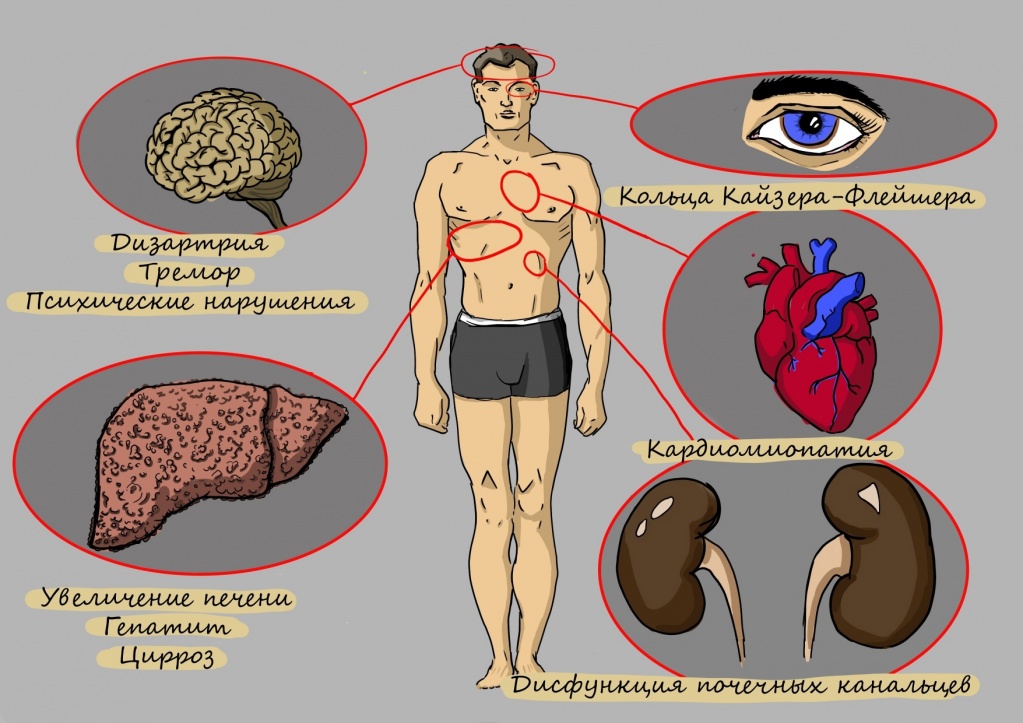
- проявления со стороны печени (гепатомегалия, острый гепатит, хронический гепатит, цирроз, фульминантная печеночная недостаточность) обычно предшествуют неврологическим и психиатрическим симптомам;
- изменение цвета кожи, слизистых оболочек и склер глаз на желтый; и живота (асцит);
- боль в верхней правой части живота;
- возникновение синяков на коже, коагулопатия;
- повышенная утомляемость;
- потеря аппетита и веса; и рвота;
- неврологические симптомы (тремор, нарушение координации и походки, дисфагия, дизартрия, хорея, спастичность, дистонические позы, мышечная ригидность);
- психические расстройства (эмоциональная нестабильность, фобии, тревожность, депрессия, компульсивное поведение, агрессивность, изменения личности и поведения) широко варьируются от пациента к пациенту, проявляются одновременно с неврологическими симптомами или развиваются в течение примерно 3 лет после их старта;
- кольца Кайзера — Флейшера на границе роговицы глаз (почти у всех пациентов с неврологическими симптомами и психическими расстройствами);
- аменорея;
- задержка полового созревания;
- выкидыши, бесплодие;
- артралгия, боль в костях;
- поражения костей и суставов (остеопороз, остеофиты);
- кардиомиопатия;
- гемолитическая анемия;
- гематурия;
- нефротический синдром;
- почечный канальцевый ацидоз;
- камни в почках;
- гепатоцеллюлярная карцинома (редко).
Диагностика
Раннее выявление болезни Вильсона — Коновалова и своевременное лечение позволяют сохранить нормальное качество жизни пациента и предотвратить опасные осложнения. Диагноз устанавливается на основании физикального осмотра, сбора жалоб, личного и семейного анамнеза пациента, лабораторной диагностики (проводятся биохимические анализы крови и мочи, которые могут показать сниженную концентрацию медь-содержащего белка церулоплазмина в плазме крови, повышение уровня печеночных ферментов, тромбоцитопению, повышенное содержание меди в моче), осмотра глаз офтальмологом с помощью щелевой лампы, КТ или МРТ головного мозга (у пациентов с неврологическими симптомами). Если лабораторные исследования не подтверждают и не исключают диагноз, врач может назначить биопсию печени, а также молекулярно-генетическое тестирование.
Болезнь Вильсона — Коновалова дифференцируют (различают) со следующими заболеваниями: вирусный гепатит, аутоиммунный гепатит, неалкогольная жировая болезнь печени, алкогольный цирроз печени, первичный склерозирующий холангит, первичный билиарный цирроз, неврологическими патологиями (эссенциальный тремор, болезнь Паркинсона с ранним началом, дистония, болезнь Хантингтона).
Лечение болезни Вильсона — Коновалова
Лекарства, воздействующего на причину болезни Вильсона — Коновалова, не существует. Пожизненное лечение направлено на снижение уровня меди в организме до нормального (нетоксичного), снижение прогрессирования заболевания и уменьшение симптомов, которые с ним связаны. Оно включает хелатирующие агенты (D-пеницилламин или триентин), которые связывают и выводят медь из организма с мочой; ацетат цинка, предотвращающий кишечную абсорбцию (всасывание) меди. Дополнительно врач может порекомендовать соблюдение диеты, исключающей продукты с повышенным содержанием меди (печень, шоколад, грибы, моллюски, орехи). Проводится динамическое наблюдение (пациент периодически сдает анализы крови и мочи) с целью оценки эффективности медикаментозной терапии.
Во время беременности лечение заболевания рекомендуется не прекращать, в этот период врач может снизить дозировку хелатирующего агента, прием ацетата цинка продолжается по стандартной схеме. Кормление грудью при использовании хелатирующего агента исключается, данных по ацетату цинка недостаточно.
При острой печеночной недостаточности, вызванной прогрессированием болезни Вильсона — Коновалова, необходима трансплантация печени.
Особенности и преимущества лечения болезни Вильсона — Коновалова в клинике Рассвет
Врачи клиники Рассвет специализируются на диагностике и лечении редких синдромов и болезней, в область их интересов в том числе входит помощь пациентам с гепатолентикулярной дегенерацией (болезнью Вильсона — Коновалова).
Наши гепатологи — врачи высокой квалификации, прекрасно подготовлены и имеют большой практический опыт. Любое редкое заболевание требует мультидисциплинарного подхода, поэтому пациент с болезнью Вильсона — Коновалова при необходимости направляется на консультацию к офтальмологу, нефрологу, психотерапевту, психиатру.
Пациентам с подозрением на болезнь Вильсона — Коновалова проводятся все необходимые диагностические исследования для подтверждения или исключения диагноза, индивидуально подбирается схема терапии.
Болезнь Вильсона ( Болезнь Вестфаля-Вильсона-Коновалова , Болезнь Вильсона-Коновалова , Гепатолентикулярная дистрофия , Гепатоцеребральная дистрофия , Лентикулярная прогрессирующая дегенерация )
Болезнь Вильсона — наследственное заболевание, передающееся по аутосомно-рецессивному типу. Возникает в условиях мутаций в гене АТР7В, кодирующем белок медьтраснпортирующей АТФазы печени. Характерный признак болезни Вильсона — накопление меди в различных органах и тканях, в большей степени в печени и базальных ганглиях. Болезнь Вильсона может протекать в брюшной, ригидно-аритмогиперкинетической, дрожательной или экстрапирамидно-корковой форме. Диагностика болезни Вильсона включает офтальмологическое обследование, биохимические анализы мочи и крови, МРТ или КТ головного мозга. Основу патогенетической терапии составляют тиоловые препараты, которые могут приниматься в течении нескольких лет и даже пожизненно.
МКБ-10
Общие сведения
Болезнь Вильсона — наследственное заболевание, передающееся по аутосомно-рецессивному типу. Возникает в условиях мутаций в гене АТР7В, кодирующем белок медьтраснпортирующей АТФ-азы печени. Характерный признак болезни Вильсона — накопление меди в различных органах и тканях, в большей степени в печени и базальных ганглиях. Первооткрыватель заболевания — А.К. Вильсон, описавший заболевание в 1912 году, в отечественной медицине — Н.А. Коновалов. Патогенез болезни Вильсона был выявлен в 1993 году. Понятию «болезнь Вильсона» соответствуют также: болезнь Вильсона-Коновалова, болезнь Вестфаля-Вильсона-Коновалова, дистрофия гепатоцеребральная, дистрофия гепатолентикулярная, дегенерация лентикулярная прогрессирующая.
Причины
Ген АТР7В картирован на длинном плече хромосомы 13 (13q14.3-q21.1). Организм человека содержит около 50-100 мг меди. Суточная потребность меди для человека — 1-2 мг. 95% абсорбированной в кишечнике меди, транспортируется в форме комплекса с церулоплазмином (один из глобулинов сыворотки, синтезируемых печенью) и только 5% в форме комплекса с альбумином. Кроме того, ион меди входит в состав важнейших метаболических ферментов (лизилоксидаза, супероксиддисмутаза, цитохром-С-оксидаза и др.). При болезни Вильсона происходит нарушение двух процессов обмена меди в печени — биосинтез главного медьсвязывающего белка (церулоплазмина) и выведение меди с желчью, следствием чего становится повышение уровня несвязанной меди в крови. Концентрация меди в различных органах (чаще всего в печени, почках, роговице и головном мозге) увеличивается, что приводит к их токсическому поражению.
Классификация
Согласно классификации Н.В. Коновалова различают пять форм болезни Вильсона:
- брюшная
- ригидно-аритмогиперкинетическая
- дрожательно-ригидная
- дрожательная
- экстрапирамидно-корковая
Для болезни Вильсона характерен клинический полиморфизм. Первые проявления заболевания могут появиться в детстве, юношестве, в зрелом возрасте и гораздо реже в зрелом возрасте. В 40-50% случаев Болезнь Вильсона манифестирует с поражения печени, в остальных — с психических и неврологических расстройств. С вовлечением в патологический процесс нервной системы обнаруживается кольцо Кайзера-Флейшера.
Брюшная форма развивается преимущественно до 40 лет. Характерный признак — тяжелое поражение печени по типу цирроза печени, хронического гепатита, фульминантного гепатита.
Ригидно-аритмогиперкинетическая форма манифестирует в детском возрасте. Начальные проявления — мышечная ригидность, амимия, смазанность речи, трудности при выполнении мелких движений, умеренное снижение интеллекта. Для этой формы заболевания характерно прогрессирующее течение, с наличием эпизодов обострения и ремиссии.
Дрожательная форма возникает в возрасте от 10 до 30 лет. Преобладающим симптомом является тремор. Кроме того, могут наблюдаться брадикинезия, брадилалия, тяжелый психоорганический синдром, эпилептические приступы.
Экстрапирамидно-корковая форма встречается весьма редко. Ее начало схоже с началом какой-либо из вышеперечисленных форм. Для нее характерны эпилептические припадки, экстрапирамидные и пирамидные нарушения и выраженный интеллектуальный дефицит.
Офтальмологическое исследование с помощью щелевой лампы выявляет кольцо Кайзера-Флейшера. Биохимические исследования мочи обнаруживают повышенную экскрецию меди в суточной моче, а также снижение концентрации церулоплазмина в крови. С помощью визуализационных методов (КТ и МРТ головного мозга) обнаруживают атрофию полушарий большого мозга и мозжечка, а также базальных ядер.
При диагностике болезни Вильсона неврологу необходимо дифференцировать ее от паркинсонизма, гепатоцеребрального синдрома, болезни Геллервордена-Шпатца. Основным дифференциально-диагностическим признаком этих заболеваний является отсутствие характерных для болезни Вильсона кольца Кайзера-Флейшера и расстройств обмена меди. Для подтверждения болезни Вильсона проводится генодиагностика.
Лечение болезни Вильсона
Основой патогенетического лечения является назначение тиоловых препаратов, в первую очередь — D-пеницилламина либо унитиола. Главное преимущество купренила — низкая токсичность и возможность длительного приема при отсутствии побочных эффектов. Его назначают по 0,15 г (1 капсула) в сутки (только после еды), в дальнейшем, в течение 2,5-3 месяцев дозу увеличивают до 6-10 капсул/сутки (оптимальная доза). Лечение D-пеницилламином проводится годами и даже пожизненно с небольшими перерывами (на 2-3 недели) в случае появления побочных эффектов (тромбоцитопения, лейкопения, обострения язвенной болезни желудка и т. д.).
Унитиол назначают в случае непереносимости (плохой переносимости) D-пеницилламина. Длительность одного курса лечения — 1 месяц, после чего лечение приостанавливают на 2,5-3 месяца. В большинстве случаев наступает улучшение общего состояния пациента, а также регресс неврологических симптомов (скованности, гиперкинезов). В случае доминирования гиперкинезов рекомендовано назначение небольших курсов нейролептиков, при ригидности — леводопы, карбидопы, тригексифенидила.
В случае тяжелого течения болезни Вильсона, при неэффективности консервативного лечения за рубежом прибегают к трансплантации печени. При положительном исходе операции состояние пациента улучшается, восстанавливается обмен меди в организме. В дальнейшем лечение пациента составляет иммуносупрессивная терапия. В России на сегодня постепенно внедряется в клиническую практику метод биогемоперфузии с изолированными живыми клетками селезенки и печени (т. н. аппарат «вспомогательная печень). Немедикаментозное лечение состоит в назначении диеты (стол №5) в целях исключения продуктов богатых медью (кофе, шоколад, бобовые, орехи и т. д.).
Прогноз и профилактика
В случае своевременного диагностирования болезни Вильсона и проведения адекватной медьснижающей терапии возможна нормализация общего состояние пациента и обмена меди в организме. Постоянный прием тиоловых препаратов по схеме, назначенной врачом-специалистом, позволяет поддерживать профессиональную и социальную активность пациента.
Для предотвращения рецидивов болезни Вильсона рекомендовано проведение лабораторных исследований крови и мочи пациента несколько раз в год. Необходим контроль следующих показателей: концентрация меди, церулоплазмина и цинка. Кроме того, рекомендовано проведение биохимического анализа крови, общего анализа крови, а также регулярные консультации у терапевта и невролога.
Медные люди: что важно знать о болезни Вильсона-Коновалова
- Вероятность встретить редкого пациента у обычного врача ничтожно мала — в России редким (орфанным)считаетсязаболевание, которое обнаруживается у одного из 10 тысяч человек. В академических аудиториях им не уделяют должного внимания, а в повседневной профессиональной практике это приводит к диагностическим ошибкам, упущенному времени и поломанным судьбам.
- Информационно-просветительский гуманитарный проект «12 месяцев» — цикл материалов о редких (орфанных) заболеваниях и жизни людей с ними — реализуют студенты и ординаторы кафедры патологической анатомии Северо-Западного государственного медицинского университета имени И. И. Мечникова (Санкт-Петербург). Они изучают генетические методы диагностики и лечения, учатся разбираться в сложных вопросах медицины и рассказывать о них, находить общий язык с редкими пациентами и их родителями, работать в мультидисциплинарной команде.
Как связаны медь, гены и болезнь Вильсона-Коновалова?
Ежедневно наш организм получает с пищей не только белки, жиры, углеводы и витамины — в него поступает множество ионов металлов и других микроэлементов, необходимых для его правильного функционирования. Например, медь, которая содержится в печени и мясе, какао и бобовых, злаках и орехах.
Несмотря на небольшую суточную потребность — всего 1,5-2,5 мг, медь участвует в обмене энергии, метаболизме железа, защите клеточных мембран — то есть практически во всех физиологических процессах в нашем теле.
Обмен меди, как и многие другие индивидуальные особенности организма генетически запрограммированы, то есть заложены в нас при рождении. Гены — это всего лишь инструкции для синтеза белков в рибосомах. Один ген — один белок, все достаточно просто. А вот какую функцию будет выполнять этот белок — зависит от его структуры.
Некоторые белки участвуют в метаболизме меди. У всех людей всасывание меди происходит в желудке и двенадцатиперстной кишке. Дальше медь транспортируется в печень, где соединяется с различными белками, затем ее часть выводится в связанном состоянии в кровь и уже после — в мочу и кал.
Чтобы меди в организме было всегда ровно столько, сколько нужно, метаболизм регулирует и выравнивает скорости ее поступления и выведения наружу.
Ключевую роль в этом уравнении играет белок-транспортер меди под названием ATP7. Он работает исправно, если в его гене, в инструкции по его сборке, нет опечаток — или мутаций, как их называют биологи. Известно уже более 800 таких опечаток в гене АТР7.
Именно из-за этих мутаций у некоторых людей белок ATP7 не работает вовсе или его функция заметно снижена. В этом случае излишки меди не выводятся из организма, а накапливаются в органах. Но много — не всегда значит хорошо. Избыток металла не дает пациентам с неработающим белком ATP7 повышенную крепкость организма, а напротив — повреждает клеточные структуры. От избытка меди прежде всего страдает головной мозг и печень.
Такую болезнь, вызванную накоплением меди в организме из-за мутации белка ATP7, называют болезнью Вильсона — по имени Сэмюэля Уилсона (Вильсона) — британского невролога, подробно описавшего симптомы заболевания в 1912 году.
В России более распространено другое название — болезнь Вильсона-Коновалова: в 1960 году советский невропатолог Николай Коновалов существенно расширил понимание болезни. Еще реже можно встретить тройное название — Вильсона-Вестфаля-Коновалова (немецкий патолог Карл Фридрих Вестфаль описал болезнь еще в 1883 году). Сами пациенты называют себя вильсонятами.
В этом ролике — краткая история открытий на пути к познанию природы этой болезни и способов помочь пациентам.
Когда появляются симптомы и какими они могут быть?
Первые симптомы болезни Вильсона-Коновалова обычно появляются на втором или третьем десятке лет жизни в виде неврологических нарушений, например, нечеткости речи, нарушении глотания, автоматических жевательных движений, и нарушений показателей работы печени.
Болезнь умеет маскироваться: она может начаться как диспепсия (боль в верхнем отделе живота) или нарушение двигательной функции и речи. И даже самый запоминающийся ее признак — медное кольцо вокруг роговицы можно обнаружить только у половины больных.
В теле не остается ни одного органа, равнодушного к нарушению обмена меди.
Но точная диагностика этого заболевания обычно представляет собой большую проблему. Биохимическое исследование показателей обмена меди в крови считается информативным, однако достоверно подтвердить мутацию можно только при помощи генетического анализа.
Чем медицина может помочь «медным людям»?
Главный на сегодняшний день способ лечения болезни Вильсона-Коновалова был предложен еще больше полувека назад — это регулярное применение препаратов, которые связывают медь и выводят ее из организма. Важным для «вильсонят» остается соблюдение строгой диеты и, при необходимости, пересадка печени.
Прерывание терапии или неправильное лечение может привести к смерти в течение нескольких месяцев. При этом медикаментозная, лекарственная терапия эффективна не для всех пациентов — побочные эффекты препаратов могут даже утяжелять состояние некоторых людей. Поэтому наука продолжает искать ответ — чем же помочь «вильсонятам»?
Не так давно исследователи научились исправлять «опечатки» в генах. В том числе — и в ATP7. Не всегда это получается хорошо, тем не менее, генная терапия может навсегда избавить пациентов с болезнью Вильсона-Коновалова от пожизненного приема препаратов и строгой диеты.
Как исследователи пытались научиться делать такую «операцию» на генах? Первые способы замены поврежденного участка гена на здоровый, в частности, применение олигонуклеотидов — коротких фрагментов ДНК или РНК, получаемых путем химического синтеза, даже у мышей приводили к едва заметному улучшению — его точно не хватило бы для помощи человеку.
Разочарования исследователей продлились до тех пор, пока они не научились «протезировать» больной ген. Для этого копию гена без поломки помещают в структуру искусственного вируса, чтобы эта неизмененная копия гена легко проникла в клетку. Освободившись от белковой оболочки, как от скафандра, ген начинает работать — и клетка наполняется правильно работающими переносчиками для меди.
Такая генная терапия представляет собой перспективную альтернативу классическому лекарственному лечению.
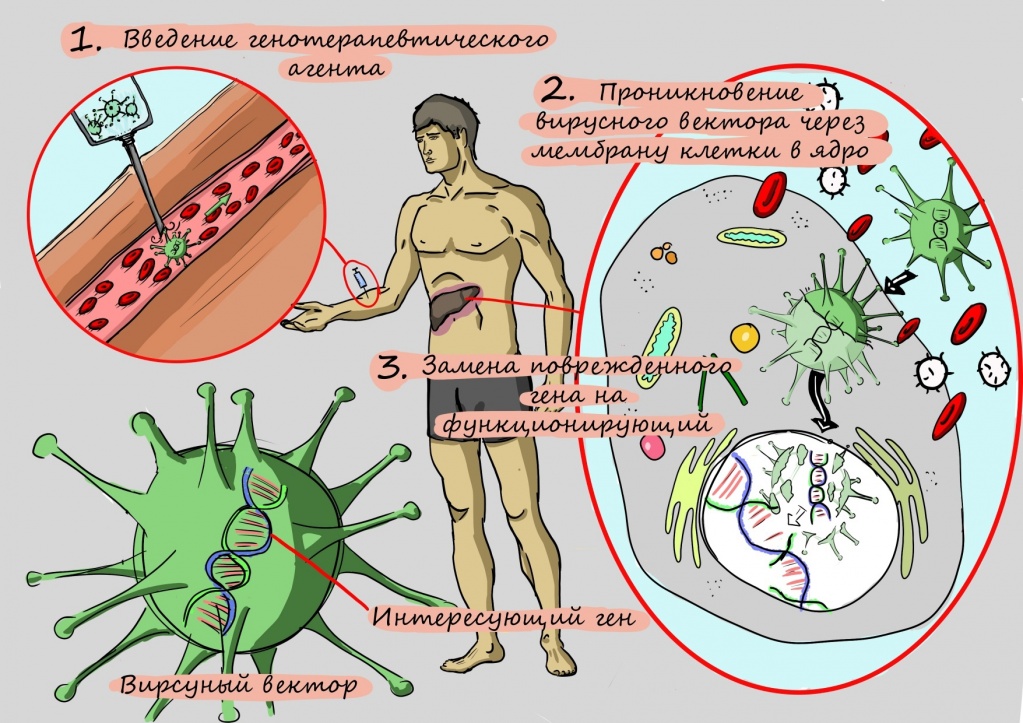
Молекулярное протезирование гена АТР7 — генная терапия
Однако при проведении этого вида терапии исследователи столкнулись с проблемой: аденовирус, который использовали для доставки гена в клетку, не может внедряться в собственный генетический аппарат клеток, — и это приводило к тому, что терапия работала недолго.
Следующим шагом в разработке терапии стало применение другого носителя — так называемого аденоассоциированного вирусного вектора.
Мышам с мутацией, которая похожа на мутацию в белке ATP7, вызывающую болезнь Вильсона-Коновалова, вводили такой вектор, и эффект оказался положительным.
Аденоассоциированный вирусный вектор имеет способность встраиваться в генетический аппарат клеток, поэтому результат лечения был более долгосрочным. Однако и этого оказалось недостаточно.
Успешный опыт подогревал ажиотаж исследователей: так, в 2021 году начались первые два клинических исследования по спасению «вильсонят» методами генной терапии (NCT04884815, NCT04537377). Через несколько лет мы узнаем — помогут ли эти разработки пациентам.
Материал подготовили: Роман Деев, Максим Пушкин, Екатерина Пичугина, Алексей Паевский, Виктория Рыжкова.
Иллюстрации Владислава Ефремова.
Болезнь Вильсона — редкое генетическое заболевание
центральной нервной системы. Чаще всего болезнь Вильсона диагностируется у подростков.
Медь играет важную роль в развитии костей, нервов, образовании коллагена, эритроцитов, меланина и многих других органов.
Медь всасывается из пищи в тонкой кишке и выводится из организма с желчью.
У людей с болезнью Вильсона медь должным образом не удаляется, а накапливается в различных тканях.
Каковы причины болезни Вильсона?
Болезнь Вильсона вызвана мутацией в гене ATP7B, который играет ключевую роль в выделении избытка меди из организма. Он наследуется
по аутосомно-рецессивному пути, что означает, что для развития заболевания необходимо наследовать мутированный ген от обоих родителей.
Каковы симптомы болезни Вильсона?
Болезнь Вильсона проявляется, когда много меди накапливается в тканях. Обычно это происходит в подростковом возрасте, но иногда может и позже.
Симптомы заболевания сильно различаются в зависимости от того, какие органы поражены болезнью.
Симптомы печени:
Неврологические симптомы — они проявляются позже всех:
- нарушенная походка;
- трудная речь – пение;
- тремор;
- ухудшение памяти;
- трудности с глотанием;
- мышечная боль;
- мышечные спазмы.
Другие симптомы, которые можно наблюдать:
- тревога, депрессия, перепады настроения;
- поведенческие изменения;
- кольцо Кайзера-Флейшера — рыжевато-коричневое кольцо вокруг радужной оболочки, появляющееся из-за отложения меди в роговице (происходит у всех пациентов с неврологическим поражением и в 1/3 случаев с поражением печени);
- нерегулярные менструации, аменорея;
- камни в почках;
- артрит;
- преждевременные менструации.
Осложнения болезни Вильсона.
Заболевание имеет прогрессирующее течение и при отсутствии лечения может привести к серьезным осложнениям: цирроз печени, печеночная
недостаточность, гемолитическая анемия, судороги, нефролитиаз.
Как диагностируется болезнь Вильсона?
Диагностика болезни Вильсона нелегка, так как симптомы возникают при других заболеваниях печени, и со временем жалобы могут измениться.
Используются следующие исследования:
- анализыкрови — определяют уровень церулоплазмина (белка, связывающего ионы меди в крови), а также уровень меди, ферментов печени;
- осмотр глаз — устанавливается кольцо Кайзера-Флейшера;
- биопсия печени;
- компьютерная томография, ядерно-магнитно-резонансная томография;
- генетическое исследование – доказывается генная мутация.
Генетическое тестирование братьев и сестер пациента является обязательным при доказательстве заболевания.
Лечение болезни Вильсона.
Крайне важно, чтобы лечение началось незамедлительно. Проводится пожизненное лечение с помощью медикаментов, которые снижают уровень меди
и останавливают прогрессирование заболевания.
Применяют: пеницилламин, триентин, который нацелен на удаление излишков меда из тканей крови, а затем выводит его из организма.
Пеницилламин имеет ряд побочных эффектов и его применение ограничено.
Ацетат цинка (гальзин) можно использовать для поддержания нормального уровня меди, что снижает кишечную абсорбцию меди. Соблюдают диету,
ограничивающую потребление продуктов с высоким содержанием меди, таких как печень, мидии, крабы, грибы, орехи, какао.
В случае серьезного повреждения печени может быть проведена трансплантация печени.
Болезнь Вильсона-Коновалова
Обращаем ваше внимание! Эта статья не является призывом к самолечению. Она написана и опубликована для повышения уровня знаний читателя о своём здоровье и понимания схемы лечения, прописанной врачом. Если вы обнаружили у себя схожие симптомы, обязательно обратитесь за помощью к доктору. Помните: самолечение может вам навредить.
Болезнь Вильсона-Коновалова – это генетическое заболевание, при котором в печени и центральной нервной системе скапливается медь.
Болезнь Вильсона-Коновалова или гепатоцеребральная дистрофия — это редкое генетически обусловленное заболевание, при котором нарушается обмен меди и в организме формируются её отложения (преимущественно в печени и центральной нервной системе).
Считается, что скрытые носители гена болезни — примерно 1% людей на Земле 1 . Болезнь наследуется по аутосомно-рецессивному типу, то есть для того, чтобы она проявилась, нужно получить дефектный ген от каждого из родителей. Именно поэтому патология встречается очень редко — в среднем 1 случай на 25 тысяч населения. Родители пациента, как правило, не знают о носительстве гена болезни. Заболевание чаще встречается в странах, где разрешены родственные браки. На сегодня известно более 200 вариантов мутаций гена, приводящих к развитию болезни Вильсона-Коновалова 2 .
Из-за дефекта гена нарушается синтез белков, осуществляющих функцию транспорта меди. В норме медь, попадающую в организм с продуктами питания, обезвреживают печеночные клетки. Дальше она выводится в желчь и покидает организм естественным путем. При болезни Вильсона-Коновалова этот механизм нарушается. На первом этапе развития медь накапливается в гепатоцитах (клетках печени), вызывая токсический гепатит.
Потом в крови появляется излишек меди: он провоцирует разрушение эритроцитов — гемолитическую анемию. Далее металл начинает откладываться в остальных органах — мозге, роговице. Наконец, из-за отравления мозга медью появляются неврологические нарушения.
Классификация и симптомы болезни Вильсона-Коновалова
В России принята классификация заболевания по клиническим формам:
- брюшная форма — преобладает поражение печени. Первые симптомы брюшной формы обычно появляются в 7–12 лет.
- аритмогиперкинетическая форма — повышенный тонус мышц, судороги, нарушения глотания и речи, снижение интеллекта, психические нарушения. Аритмогиперкинетическая форма, как правило, проявляется в возрасте от 7 до 15 лет.
- дрожательно-ригидная форма. Считается относительно доброкачественной. Преобладает тремор конечностей, ригидность мышц. Выраженность проявлений со стороны внутренних органов и психических нарушений варьирует. Дебют дрожательно-ригидной формы обычно происходит в возрасте от 15 до 25 лет.
- дрожательная форма. Считается одной из самых благоприятных. Тонус мышц не изменен. Выраженное дрожание конечностей. Интеллект сохранен, проявления со стороны внутренних органов минимальны. Дрожательная форма может проявиться в 20-30 и даже 50 лет.
- экстрапирамидно-корковая форма. Преобладают двигательные и психические нарушения: центральные парезы, эпиприступы, быстрое снижение интеллекта и психиатрические проявления.
Болезнь может возникнуть в любом возрасте, но чаще всего ее дебют приходится на 11–25 лет. Чем позже появляются симптомы, тем медленней развивается заболевание. Описанные в литературе формы редко встречаются «в чистом» виде. Как правило, отмечаются симптомы со стороны практически всех органов и систем.
Со стороны печени болезнь Вильсона-Коновалова обычно проявляется в виде острого или хронического гепатита с исходом в цирроз печени и портальную гипертензию:
- ;
- повышение температуры;
- тошнота, рвота; ; ;
- на поздних стадиях — отеки, асцит, кровотечения (проявления цирроза).
Со стороны нервной системы отмечаются:
- тремор (дрожание) конечностей;
- нарушения походки;
- повышенное слюноотделение;
- маскообразное лицо;
- эпилептические приступы;
- ригидность («застывание») мышц — больные нередко замирают в самых причудливых позах.
Со стороны психики:
- аффективные вспышки;
- снижение интеллекта;
- изменения личности, «дурашливость»;
- галлюцинации, психозы.
Со стороны системы крови:
- гемолиз (распад эритроцитов);
- анемия;
- тромбоцитопения;
- нарушения свертываемости крови.
Со стороны почек:
- повышение уровня креатинина крови;
- отеки;
- микрогематурия (скрытая кровь в моче);
- протеинурия;
- нефролитиаз (камни).
Со стороны эндокринной системы:
- задержка полового созревания;
- гинекомастия;
- гирсутизм; ;
- аменорея;
- спонтанные аборты.
Таким образом, клиническая картина складывается сложная и крайне разнообразная, что значительно затрудняет диагностику заболевания.
Диагностика болезни
Нередко болезнь Вильсона-Коновалова диагностируется только на стадии выраженного цирроза. Это связано со сложностями диагностики. Скрининговый тест на заболевание — снижение церулоплазмина в сыворотке крови — может дать как ложноположительные (при циррозе печени другого происхождения, нефротическом синдроме, белковом голодании, мальадсорбции), так и ложноотрицательные (при активном воспалении в печени, приеме гормональных контрацептивов, во время беременности, при рассеянном склерозе) результаты.
Диагностически значимым может быть небольшое снижение количества меди в сыворотке крови и многократное превышение ее нормального количества в моче (более 100 мкг/сут при норме до 40 мкг/сут), но при бессимптомном течении заболевания и эти показатели могут оставаться в пределах нормы.
Поэтому в сомнительных случаях проводят биопсию печени и генетический анализ на наличие дефектного гена.
Традиционные клинические и биохимические анализы крови, как правило, не показательны: возможны анемия, тромбоцитопения, повышение АЛТ, АСТ и билирубина, снижение альбуминов и протромбинового индекса. Но все эти признаки характерны для большинства заболеваний печени, а не только для болезни Вильсона-Коновалова.
Лечение болезни Вильсона-Коновалова
Рекомендуется вариант диеты №5 с низким содержанием меди. Пить необходимо только дистиллированную воду.
Для детоксикации назначают препарат, связывающий тяжелые металлы, в том числе медь (D-пеницилламин или триентин). В первые недели терапии состояние пациента может ухудшаться, так как лекарство выводит медь из печени и на некоторое время ее концентрация в крови повышается. Постепенно больному становится лучше.
Принимать препарат для удаления излишков меди из организма необходимо пожизненно, перерывы могут спровоцировать рецидив болезни с развитием острой печеночной недостаточности. Важно: лекарство связывает не только медь, но и витамин В6, поэтому его недостаток нужно восполнять.
Для поддержания функции печени рекомендованы препараты на основе урсодезоксихолевой кислоты (урсосан). Она защищает орган от токсического влияния меди, улучшает качество желчи и её отток. При болезни Вильсона-Коновалова необходимы периодические курсовые приемы УДХК. Дозировку лекарства и количество таких курсов в год нужно уточнять у врача-гастроэнтеролога.
В тяжелых случаях необходима пересадка печени.
При ранней диагностике и своевременно начатом лечении прогноз благоприятный — продолжительность жизни пациентов не отличается от средней в популяции. При поздней диагностике прогноз резко ухудшается, так как изменения со стороны печени необратимы.
Профилактика заключается в генетическом консультировании перед планированием беременности.
1 Нарушения обмена меди (болезнь Вильсона) у детей. Федеральные клинические рекомендации, 2016.
2 Е.Ю. Ерёмина. Болезнь Вильсона-Коновалова. Вестник современной клинической медицины, 2011.
Обращаем ваше внимание! Эта статья не является призывом к самолечению. Она написана и опубликована для повышения уровня знаний читателя о своём здоровье и понимания схемы лечения, прописанной врачом. Помните: самолечение может вам навредить. Если вы обнаружили у себя схожие симптомы, обязательно обратитесь за помощью к доктору:

Читайте также: